Molecular Dynamics Simulation¶
This tutorial will guide the user through the general workflow to perform a molecular dynamicsi (MD) simulation for a water dimer by using python based packages such as eDFTpy and the Atomic Simulation Environment (ASE)
Note
To avoid mixing some files, we strongly recommend setup each calculation (e.g. Optimization of density, relaxation of a structure of an MD simulation) in separate folders.
Input Files¶
Same as the relaxation step, MD simulation also, requires a set of input files divided into four different types:
Global system coordinates
eDFTpy input file
Aditional subsystem configuration
ASE input file
Global system coordinates¶
In this tutorial, we are going to use the optimized structure obtaned as an output from the relaxation of a structure tutorial opt.xyz however, this workflow in real conditions depends on the needs of each user, this means there may be times when the user does not have to perform a relaxation calculation prior to the molecular dynamics simulation.
opt.xyz
8
Lattice="12.0 0.0 0.0 0.0 12.0 0.0 0.0 0.0 12.0" Properties=species:S:1:pos:R:3:forces:R:3 energy=-2694.8624247979765 stress="0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0" pbc="T T T"
Na 3.86470766 3.72085638 4.21569556 -0.00186095 -0.01499023 0.00394566
Cl 5.43080951 6.96996202 7.28173141 -0.00318058 0.02479020 0.01575104
O 2.52019224 2.60825963 2.67929060 0.00143356 -0.00170748 -0.01002334
H 2.08539043 2.99876243 1.89264120 -0.00729920 0.00849589 -0.00562617
H 2.31711235 1.65197643 2.61433000 -0.00410939 -0.01766387 0.01191952
O 5.11120809 5.01245357 5.42830940 0.01606948 0.00478632 -0.03197085
H 4.80166277 5.40513659 6.29470526 0.00205590 0.00250708 -0.00322683
H 5.86891694 5.63259297 5.32363258 -0.00310882 -0.00621791 0.01923098
eDFTpy input file¶
Essentially, this file is the same as the one provided in the previous step. Nevertheless, for this step of the tutorial, you should change the cell file name to opt.xyz. Also, we are going to use the adaptive definition of the subsystems for the global system through the MD simulation, using the same distance method introduced in the SCF tutorial with the automatic definition of subsystems. It means we should include the decompose keyword and all of the atomic radius in the [GSYSTEM] flag, as follows:
input.ini
[JOB]
task = Optdensity
[PATH]
pp = ./
cell = ./
[PP]
O = o_ONCV_PBE-1.2.upf
H = h_ONCV_PBE-1.2.upf
Na = na_pbe_v1.5.uspp.F.UPF
Cl = cl_pbe_v1.4.uspp.F.UPF
[OPT]
maxiter = 200
econv = 1e-6
[MOL]
charge-Na = 1
charge-H2O = 0
charge-Cl = -1
[GSYSTEM]
cell-file = opt.xyz
grid-ecut = 1200
exc-x_str = gga_x_pbe
exc-c_str = gga_c_pbe
kedf-kedf = GGA
kedf-k_str = revAPBEK
decompose-method = distance
decompose-radius-O = 0.90
decompose-radius-H = 0.60
decompose-radius-Cl = 1.0
decompose-radius-Na = 0.6
[SUB_NA]
calculator = qe
embed = KE XC
cell-split = 0.5 0.5 0.5
cell-index = 0:1
grid-ecut = 2400
mix-coef = 0.7
[SUB_CL]
calculator = qe
embed = KE XC
cell-split = 0.5 0.5 0.5
cell-index = 1:2
basefile = qe_in_cl.in
density-use_gaussians = True
mix-coef = 0.7
[SUB_H2O]
calculator = qe
embed = KE XC
cell-split = 0.5 0.5 0.5
cell-index = 2:
decompose-method = distance
decompose-radius-O = 0.90
decompose-radius-H = 0.60
grid-ecut = 2400
mix-coef = 0.7
cell-file = opt.xyz
kedf-k_str = revAPBEK
decompose-method = distance
decompose-radius-O = 0.90
decompose-radius-H = 0.60
decompose-radius-Cl = 1.0
Aditional subsystem configuration¶
We use the same base file provided for the relaxation step, you can change to different parameters if you require.
qe_in_cl.in
&SYSTEM
ecutwfc = 40
ecutrho = 400
nosym = .true.
occupations = 'smearing'
degauss = 0.001
smearing = 'gaussian'
/
&ELECTRONS
/
ASE input file¶
Followed the ASE input file explanation for the relaxation of a strcutre, this input file also is written following the ASE input file format. Contrary to the previous file, this input file includes definitions of temperature, thermodynamics ensemble, thermostat, time step, etc used to run the simulation, follows we summarize the ones that we consider you should think about before setting up a real calculation. Check ASE Molecular dynamics module for an extensive keywords description.
ase_nvt.py
#!/usr/bin/env python3
import os
from edftpy.api.api4ase import eDFTpyCalculator
from edftpy.config import read_conf
from edftpy.interface import conf2init
from edftpy.mpi import sprint, pmi
############################## initial ##############################
conf = read_conf('./input.ini')
graphtopo = conf2init(conf, pmi.size > 0)
cell_file = conf["PATH"]["cell"] +os.sep+ conf['GSYSTEM']["cell"]["file"]
#-----------------------------------------------------------------------
import ase.io
import ase.md
from ase.io.trajectory import Trajectory
from ase import units
from ase.md.npt import NPT
from ase.md.langevin import Langevin
from ase.md.nvtberendsen import NVTBerendsen
from ase.md.andersen import Andersen
from ase.md.velocitydistribution import MaxwellBoltzmannDistribution
atoms = ase.io.read(cell_file)
T = 300 # Kelvin
# T *= units.kB
calc = eDFTpyCalculator(config = conf, graphtopo = graphtopo)
atoms.set_calculator(calc)
MaxwellBoltzmannDistribution(atoms, temperature_K = T, force_temp=True)
#dyn = Andersen(atoms, 1.5 * units.fs, temperature_K = T, andersen_prob=0.02)
dyn = NVTBerendsen(atoms, 0.7 * units.fs, temperature_K = T, taut=0.2*1000*units.fs)
step = 0
interval = 1
def printenergy(a=atoms):
global step, interval
epot = a.get_potential_energy() / len(a)
ekin = a.get_kinetic_energy() / len(a)
sprint(
"Step={:<8d} Epot={:.5f} Ekin={:.5f} T={:.3f} Etot={:.5f}".format(
step, epot, ekin, ekin / (0.7 * units.kB), epot + ekin
)
)
step += interval
dyn.attach(printenergy, interval=1)
traj = Trajectory("md.traj", "w", atoms)
dyn.attach(traj.write, interval=1)
dyn.attach(traj.write, interval=1)
nsteps = 20
# dyn.run(nsteps)
from ase.optimize.optimize import Dynamics
dyn.max_steps = dyn.nsteps + nsteps
for converged in Dynamics.irun(dyn):
if os.path.isfile('edftpy_stopfile'): exit()
atoms = ase.io.read(cell_file) # Name of the file from ASE will read the coordinates
T = 300 # Base temperature at the MD simulation will be runnied.
MaxwellBoltzmannDistribution(atoms, temperature_K = T, force_temp=True) # Initialize the velocities at the defined temperature base on the Maxwell Boltzmann distribution
dyn = NVTBerendsen(atoms, 0.7 * units.fs, temperature_K = T, taut=0.2*1000*units.fs) # Define the type of thermostat we will used for the simulation and the time step used, for this tutorial we used a dt = 0.7 fs.
traj = Trajectory("md.traj", "w", atoms) # Name of the output trajectory file
nsteps = 10 # Number of steps you will run your simulation
Running eDFTpy¶
Once you finish setting out the calculation, you can run the calculation using:
$ mpirun -n 4 python -m ase_relax.py > log
Note
Here the log file storages the summary of the calculation.
Warning
If you are using python3 version you should change all the python command lines wrote here from python to python3
Additional output Files¶
edftpy_gsystem.xyz Global system coordinates for the last step of the simulation
md.traj Output trajectory file for the global system.
Preliminary Analysis¶
Once you finished your simulation you would like to analyze your results, most of the time your structural analysis will require your trajectory file (md.traj). Although this trajectory formatis only useful if you are using the ASE package to perform visual analysis.
Open the trajectory file with the ASE visualization tool by typing:
$ ase gui md.traj
Once you open MD trajectory file observe the step by step structure change and a graphic generator which includes a set of graphics check ASE’s GUI. Here we show the variation of the total energy (eV) in function of the number steps for the MD simulation of the binary salt (NaCl) molecule surrounded by a water dimer by 20 steps of simulation.
|
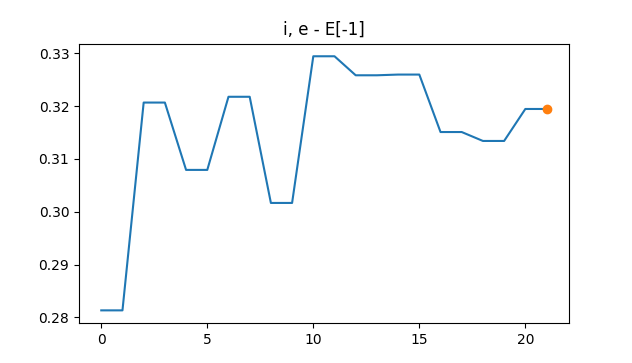
Note
For the purposes of this tutorial, the configured input file only includes 20 steps, otherwise the wait time would be proportional to a couple of days using four (4) cores.
Convert the format of a trajectory file¶
If you would like to use any other package to carry out structural analysis will be useful for you to covert this trajectory file to a most common type as xyz. We developed a python script that you can use to convert your trajectory files by typing:
$ python -m edftpy –-convert –-traj md.traj -o md-traj.xyz
You can check the command line miscellaneous for eDFTpy in the command line section of the sofware website.
Restarting a MD simulation¶
Restarting a calculation could be a powerful option taking into account the time-consuming disadvantage of an ab-initio molecular dynamics simulation. Here we give the user a guide on how can restart an MD simulation
To restart a simulation the user should modify the eDFTpy and ASE input files either Structure optimizartion or MD production.
Copy your trajectory file (md.traj) to a new file:
$ cp md.traj md-1.traj
Create a new global system coordinate file using the last geometry coordinates:
$ cp edftpy_gsystem.xyz rst-1.xyz
Change the file name for the cell-file in the eDFTpy input file
from
cell-file = opt.xyz
to
cell-file = rst-1.xyz
Change the cell_file in the ASE input file to your old trajectory file
from
atoms = ase.io.read(cell_file)
to
atoms = ase.io.read("md-1.traj")
As you already initialize velocities from your las trajectory you should not include the Maxwell Boltzmann Distribution line, you can comment this line adding a hash # at he begining of the line
To avoid to concatenate all the trajectory file at the end of the simulation you can attempt the new trajectory to the old md.traj file modifying the trajectory file section:
from
traj = Trajectory("md.traj", "w", atoms)
to
traj = Trajectory("md.traj", "a", atoms)
Restart your simulation using:
$ mpirun -n 4 python -m ase_relax.py > log